University of Missouri
Computational RNA Biophysics
RNA 2D structure folding
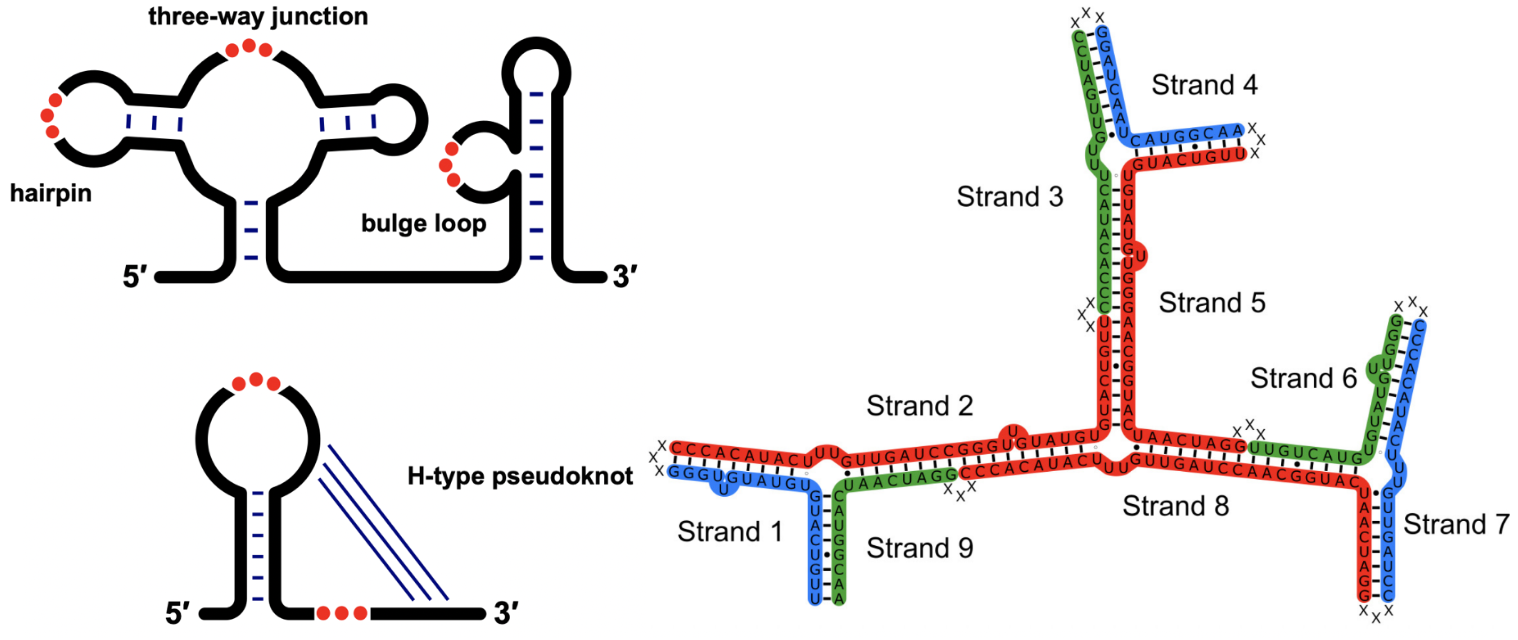
The RNA folding process is often hierarchical, beginning with the formation of RNA secondary (2D) structures through base pairing. The 2D structure prediction models are hampered by the availability of thermodynamic parameters of RNA motifs. We have addressed this challenge and developed two models
• Vfold2D [1-4] a physics-based model that employs the statistics of known structures to compute energy parameters, enabling the prediction of 2D structures with pseudoknots and non-canonical base pairs
• VfoldMCPX [5-6] a physics-based model designed to predict the structure of multi-strand RNA assembly.
These models significantly enhance the predictive capability of RNA 2D folding predictions and exhibit improved prediction accuracy.
- Y. Cheng, S. Zhang, X. Xu, and S. J. Chen. Vfold2D-MC: a physics-based hybrid model for predicting RNA secondary structure folding. J. Phys. Chem. B, 125, 10108-10118, (2021).
- S. Cao, and S. J. Chen. Predicting structures and stabilities for H-type pseudoknots with interhelix loops. RNA, 15, 696-706, (2009).
- S. Cao, and S. J. Chen. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res., 34, 2634-2652, (2006).
- S. Cao, and S. J. Chen. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA, 11, 1884-1897, (2005).
- S. Zhang, Y. Cheng, P. Guo, S. J. Chen. VfoldMCPX: predicting multistrand RNA complexes. RNA, 28, 596-608, (2022).
- X. Xu, and S. J. Chen. VfoldCPX server: predicting RNA-RNA complex structure and stability. PLoS One, 11, e0163454, (2016).
RNA 3D structure folding
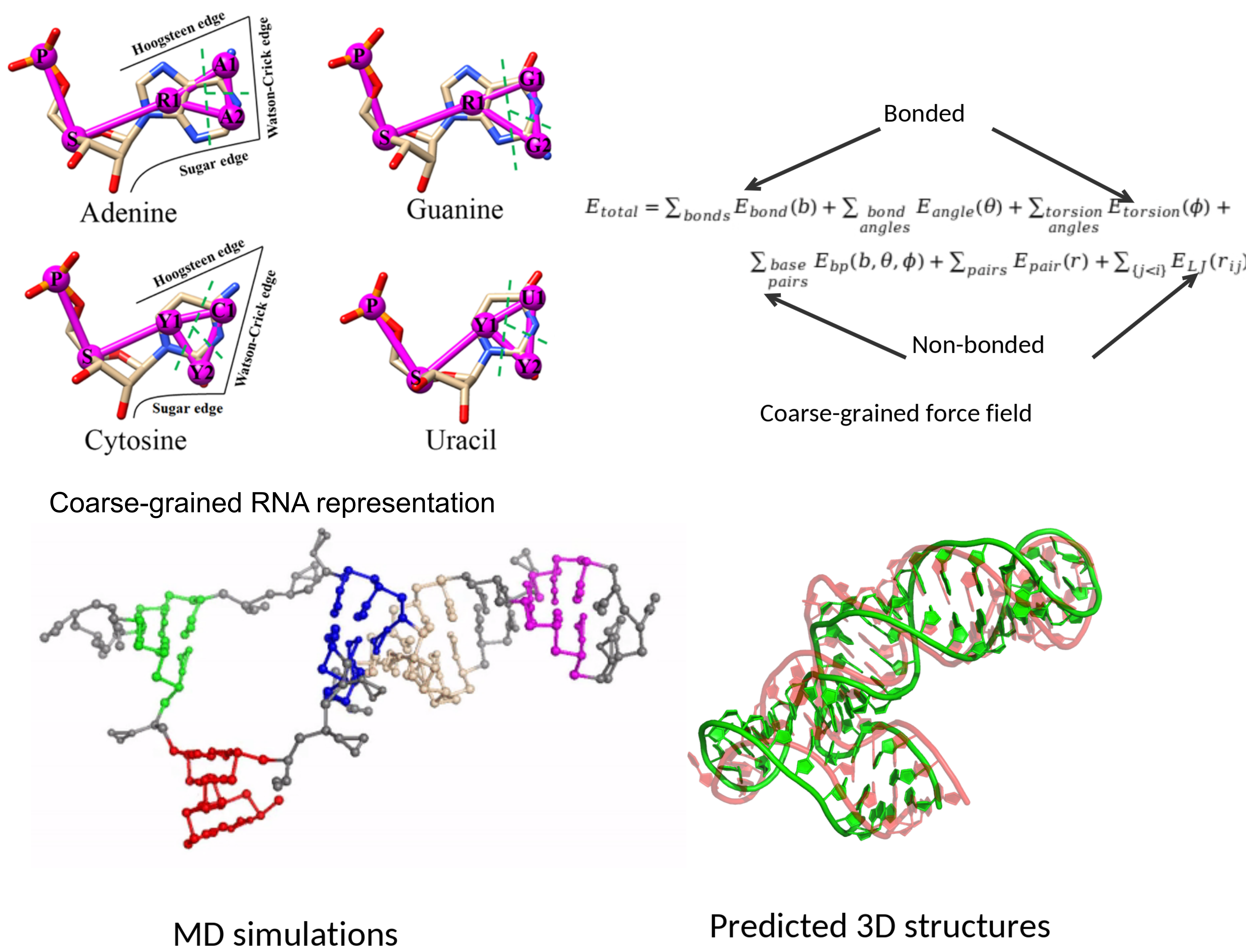
Modeling the three-dimensional (3D) structure of RNA is crucial for understanding RNA functions and for the design of RNA-based drugs and therapeutics. Our research group addresses this challenge by developing multiple RNA 3D structure prediction models, including
• Vfold3D [1] a motif template-based model• VfoldLA [2] a loop template-based model
• IsRNA [3-5], a model based on statistical potentials and coarse-grained molecular dynamics
Additionally, we developed Vfold-Pipeline [6], an end-to-end, fully automated solution for RNA 3D structure modeling from a given sequence.
These models adopt various methods and perspectives to tackle the complexities of RNA 3D folding, thereby enhancing the accuracy of RNA structure predictions.
- S. Cao, and S. J. Chen. Physics-based de novo prediction of RNA 3D structures. J. Phys. Chem. B, 115(14), 4216-4226, (2011)
- X. Xu, and S. J. Chen. Hierarchical assembly of RNA three-dimensional structures based on loop templates. J. Phys. Chem. B, 122(21), 5327-5335, (2017).
- D. Zhang, and S. J. Chen. IsRNA: An iterative simulated reference state approach to modeling correlated interactions in RNA folding. J. Chem. Theory Comput., 14(4), 2230-2239, (2018).
- D. Zhang, J. Li, and S. J. Chen. IsRNA1: de novo prediction and blind screening of RNA 3D structures. J. Chem. Theory Comput., 17(3), 1842-1857, (2021).
- D. Zhang, S. J. Chen, and R. Zhou, Modeling noncanonical RNA base pairs by a coarse-grained IsRNA2 model. J. Phys. Chem. B, 125(43), 11907-11915, (2021).
- J. Li, S. Zhang, D. Zhang, and S. J. Chen Vfold-Pipeline: a web server for RNA 3D structure prediction from sequences. Bioinform., 38(16), 4042-4043, (2022).
RNA-ligand docking
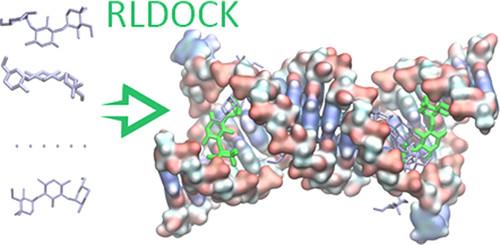
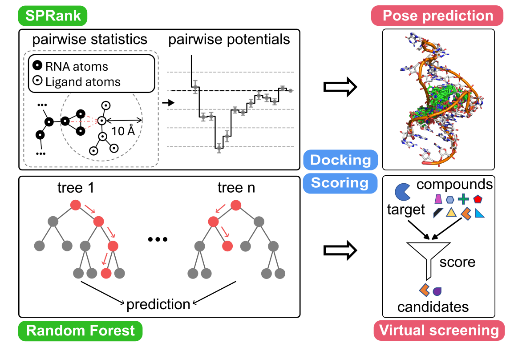
Identifying small molecules that bind RNA targets can lead to new therapeutics for diseases involving dysregulated RNA functions. Effective computational screening in this field demands robust docking software and scoring functions to predict binding modes and enable virtual screening. Our group recently developed two new tools
• RLDOCK [1,2] offers an exhaustive sampling of binding sites and an accurate energy scoring
• SPRank [3] a hybrid machine-learning and knowledge-based scoring function, enhances docking tools by sampling flexible ligands against diverse RNA structures.
Both models have shown improved binding mode prediction and virtual screening across multiple RNA targets such as HIV-1 TAR, underscoring their potential in RNA-targeted drug discovery.
- L. Z. Sun, Y. W. Jiang, Y. Z. Zhou, and S. J. Chen, RLDOCK: A new method for predicting RNA-ligand interactions. J. Chem. Theory Comput., 16, 7173-7183, (2020).
- Y. W. Jiang, and S. J. Chen, RLDOCK method for predicting RNA-small molecule binding modes. Methods., 197, 97-105, (2022).
- Y. Z. Zhou, Y. W. Jiang, and S. J. Chen, SPRank - A knowledge-based scoring function for RNA-ligand pose prediction and virtual screening. J. Chem. Theory Comput., 20, 7358-7369, (2004).
CRISPR-Cas9 system
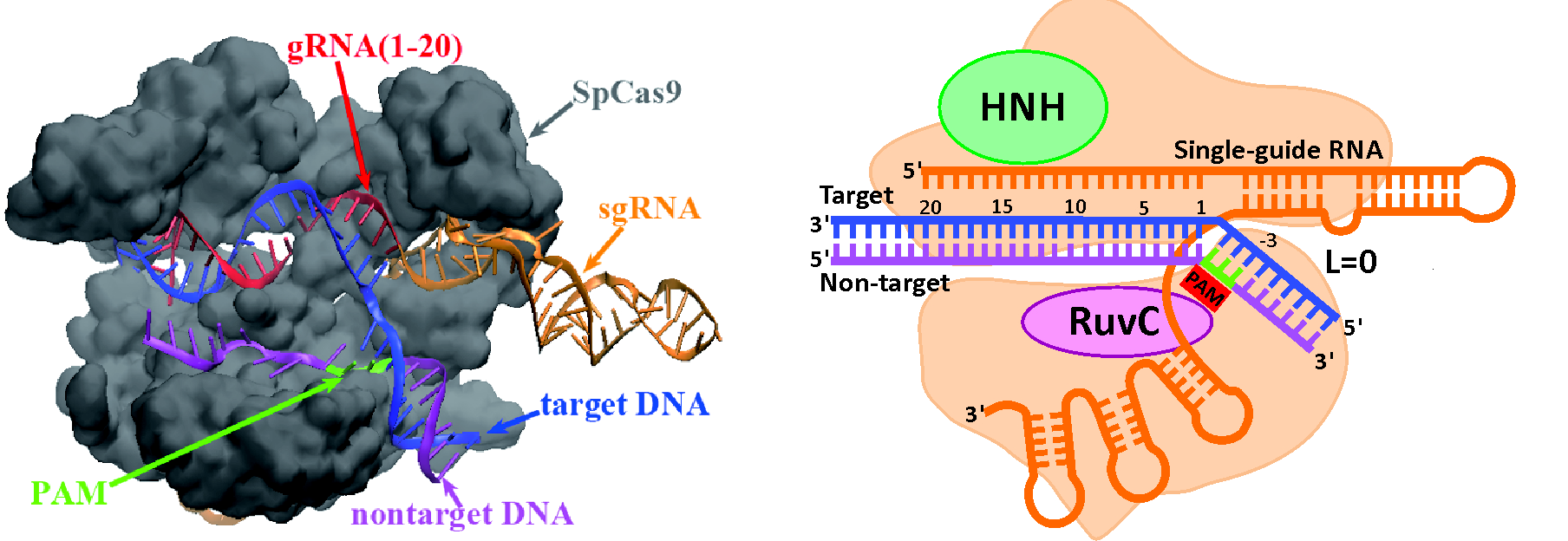
The CRISPR/Cas9 nuclease system is a transformative technology for genome engineering, and its success hinges on the strategic design of guide RNAs (gRNAs) to precisely control on- and off-target cleavage efficiencies. To address this challenge, we developed two computational models built on the underlying physical principles of CRISPR gene editing.
• uCRISPR [1] model leverages integrated energy parameters to evaluate R-loop folding stability.
• kCRISPR [2] model applies kinetic Monte Carlo simulations to capture R-loop folding/unfolding kinetics and employs a supervised ensemble learning algorithm to pinpoint key determinants of cleavage efficiency.
Together, these models provide promising tools to accurately predict and optimize gRNA performance at both on- and off-target sites.
- Zhang D, Hurst T, Duan D, Chen SJ. Unified energetics analysis unravels SpCas9 cleavage activity for optimal gRNA design. Proceedings of the National Academy of Sciences. 2019 Apr 30;116(18):8693-8.
- Xu, X, Duan, DS, Chen, SJ. (2017) CRISPR-Cas9 cleavage efficiency correlates strongly with target-sgRNA folding stability: from physical mechanism to off-target assessment.Sci Rep. 7(1):143. doi: 10.1038/s41598-017-00180-1.
RNA deep learning
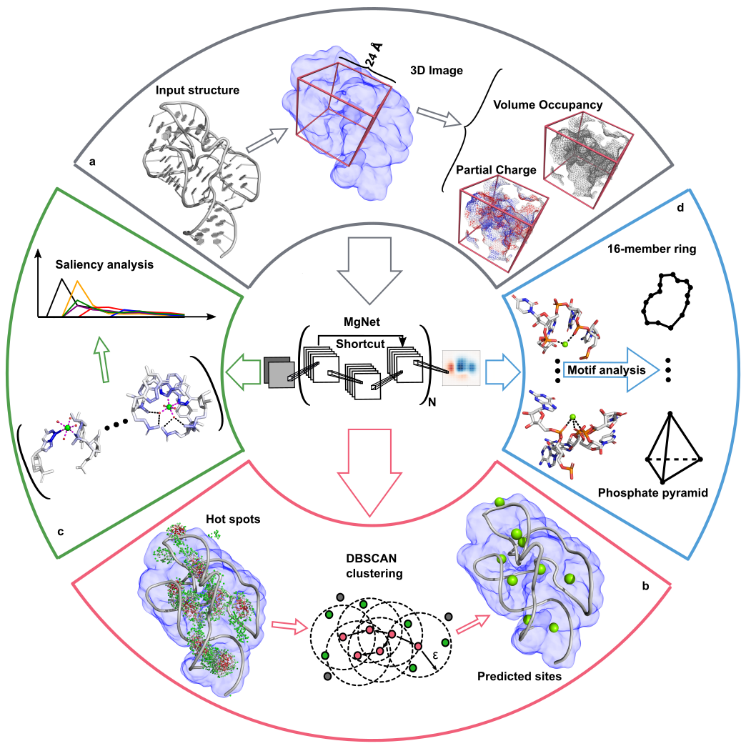
Metal ions are critical for RNA structure and function, but accurately modeling metal ion (e.g., Mg²⁺) effects remains a major challenge. Our group previously developed statistical mechanics-based models to account for ion correlation and fluctuations in nucleic acids: TBI [1] and MCTBI [2,3].
Building on this, we recently developed a deep learning method, MgNet which treats RNA-ion complexes as images and predicts Mg²⁺ binding sites that are tightly coordinated with RNA atoms. MgNet identifies critical atoms for inner/outer-sphere coordination and uncovers new ion-binding motifs. This model can complement X-ray crystallography for precise metal ion site identification.
- Z. J. Tan, and S. J. Chen, Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. J. Chem. Phys., 122, 44903, (2005).
- L. Z. Sun, and S. J. Chen, Monte Carlo Tightly Bound Ion Model: Predicting Ion-Binding Properties of RNA with Ion Correlations and Fluctuations. J. Chem. Theory Comput., 12, 3370–3381, (2016).
- L. Z. Sun, J. X. Zhang, and S. J. Chen, MCTBI: a web server for predicting metal ion effects in RNA structures. RNA., 23, 1155–1165, (2017).
- Y. Z. Zhou, and S. J. Chen, Graph deep learning locates magnesium ions in RNA. QRB Discov., 3, E20, (2022).